喀拉昆仑南部熊彩岗日地区冰川细菌多样性研究
【类型】期刊
【作者】张淑红,包格日乐,李治国,陈红歌(河南农业大学生命科学学院;商丘师范学院生命科学学院;商丘师范学院环境与规划学院)
【作者单位】河南农业大学生命科学学院;商丘师范学院生命科学学院;商丘师范学院环境与规划学院
【刊名】河南农业大学学报
【关键词】 细菌;多样性;冰川;熊彩岗日地区
【资助项】国家自然科学基金项目(31100369,41101072,41330526)
【ISSN号】1000-2340
【页码】P275-281
【年份】2019
【期号】第2期
【期刊卷】1;|7;|8;|4;|5
【摘要】通过纯培养和高通量测序,对喀拉昆仑南部熊彩岗日地区冰川雪样和水样细菌多样性进行了研究。纯培养结果表明,冰川细菌由Bacteroidetes、Actinobacteria、Deinococcus-Thermus、Proteobacteria4个门组成,属水平由Pseudomonas、Janthinobacterium、Flavobacterium、Devosia、Duganella、Deinococcus、Cryobacterium、Rugamonas、Arthrobacter、Mycetocola、Massilia、Salinibacterium组成;高通量测序结果表明,冰川细菌主要由Proteobacteria、Bacteroidetes、Firmicutes 3个门组成,由Acidovorax、Bacillus、Bacteroides、Brevundimonas、Herbaspirillum、Massilia、Pedobacter、Phenylobacterium、Polaromonas、Pseudorhodobacter、Variovorax等属组成。2种方法综合起来更能全面地分析细菌群落信息。雪环境和水环境的细菌多样性差异不大。细菌群落组成既有重复的,也有各自环境所特有的。同时也检测到了熊彩岗日地区冰川所特有的细菌类群。
【全文】 文献传递
喀拉昆仑南部熊彩岗日地区冰川细菌多样性研究
摘要:通过纯培养和高通量测序,对喀拉昆仑南部熊彩岗日地区冰川雪样和水样细菌多样性进行了研究。纯培养结果表明,冰川细菌由Bacteroidetes、Actinobacteria、Deinococcus-Thermus、Proteobacteria4个门组成,属水平由Pseudomonas、Janthinobacterium、Flavobacterium、Devosia、Duganella、Deinococcus、Cryobacterium、Rugamonas、Arthrobacter、Mycetocola、Massilia、Salinibacterium组成;高通量测序结果表明,冰川细菌主要由Proteobacteria、Bacteroidetes、Firmicutes 3个门组成,由Acidovorax、Bacillus、Bacteroides、Brevundimonas、Herbaspirillum、Massilia、Pedobacter、Phenylobacterium、Polaromonas、Pseudorhodobacter、Variovorax等属组成。2种方法综合起来更能全面地分析细菌群落信息。雪环境和水环境的细菌多样性差异不大。细菌群落组成既有重复的,也有各自环境所特有的。同时也检测到了熊彩岗日地区冰川所特有的细菌类群。
关键词:细菌;多样性;冰川;熊彩岗日地区
冰川,由于其独特的地理环境特征,蕴藏着大量具有独特遗传学和适应环境变化机制的微生物,是一个天然的微生物“储存库”,记录着不同时期大气环流向冰川输送的微生物菌群数量和结构等信息,是包含生物进化以及地球上生物生存环境变化信息的优良介质[1]。以冰芯和雪样中微生物为研究对象的工作诸多,而且,随着近年来冰川退缩的不断加剧,关于冰川退缩前沿裸露地微生物群落演替特点受到了诸多关注。而对于退缩过程中形成的不同生境之间微生物群落差异的研究较少,仅见到了老虎沟12号冰川的报道[2,3],而且运用高通量测序技术研究冰川微生物的工作才刚刚开始。本研究通过对尚未开展过冰川微生物研究工作的熊彩岗日地区的细菌进行纯培养及高通量测序分析,采集了不同海拔处的高位点雪、消融区雪、冰川融水及冰川末端河水样品。一方面分析不同冰川环境细菌群落的差异,另一方面将熊彩岗日地区冰川的菌群结构与其它冰川位点进行比较,分析熊彩岗日地区冰川细菌群落所独有的特征。
1 材料与方法
1.1 样品的采集
喀拉昆仑山位于青藏高原西北侧,其主体部分在新疆维吾尔自治区与克什米尔的交界线上。狮泉河气象站(32°30′N,80°08′E,2 924 m)在1961—2013年的观测结果是,年均温度为0.71 ℃,年均沉积物厚度为70.60 mm。本试验采样地点为喀喇昆仑南部的熊彩岗日地区,1968—2013年该地区冰川总面积减少了2.63 km2,退缩面积占1968年此地区冰川总面积的1.45%[4]。
2013年7月,在喀拉昆仑南部熊彩岗日地区共采集4个样品:高位点雪样(34°15′11.70″N,80°17′19.40″E,5 881 m)、消融区雪(34°15′11.36″N,80°17′32.89″E,5 731.21 m)、冰川融水(34°15′11.28″N,80°17′32.89″E,5 730.45 m)、河水(34°15′14.10″N,80°17′33.60″E,5 704.79 m)。每种样品3个重复。试验样品低温转运,4 ℃保存备用。
1.2 可培养细菌的纯培养
1.2.1 培养基 R2A培养基: 蛋白胨0.5 g·L-1,酵母粉0.5 g·L-1,胰蛋白胨0.5 g·L-1,酸水解酪蛋白0.5 g·L-1,葡萄糖0.5 g·L-1,可溶性淀粉0.5 g·L-1,磷酸氢二钾0.3 g·L-1,丙酮酸钠0.3 g·L-1,七水硫酸镁0.05 g·L-1,琼脂15 g·L-1,pH值为7.2~7.4。
LB培养基:胰蛋白胨10 g·L-1,酵母提取物5 g·L-1,氯化钠10 g·L-1,琼脂15 g·L-1,pH值为7.0~7.2。
TSA培养基:胰蛋白胨15 g·L-1,大豆胨5 g·L-1,氯化钠5 g·L-1,琼脂15 g·L-1,pH值为7.0~7.4。
1.2.2 分离与纯化 用R2A、0.25R2A、0.2LB、0.5TSA 4种培养基进行纯培养。在无菌条件下,每种样品分别吸取100 μL,涂布于4种培养基上。每种培养基涂6个平板,3个置于4 ℃培养15 d,另外3个置于15 ℃培养7 d。形成肉眼可见的菌落后,依据细菌形态特征进行分离与纯化,统计同一形态菌落数及总菌落数。纯化后的细菌于4 ℃斜面保存及30%甘油-20 ℃保藏备用。
1.2.3 恢复培养的菌株DNA提取 参照ZHOU等[5]方法提取细菌DNA并做了一些改动。具体过程如下:(1)吸取菌液于2 mL离心管中,4 000 r·min-1离心10 min收集菌体。(2)菌体中加入1 mL 磷酸缓冲液PBS(KCl 0.2 g·L-1,KH2PO4 0.24 g·L-1,NaCl 8 g·L-1,Na2HPO4 1.44 g·L-1)混匀,12 000 r·min-1离心4 min,此过程为洗菌。(3)弃上清液,加入650 μL 提取液(100 mmol Tris-Cl,100 mmol Na2-EDTA,100 mmol Na2HPO4,1.5 mmol NaCl,1% CTAB),10 g·L-1蛋白酶K和10 g·L-1溶菌酶各10 μL,气浴振荡器37 ℃振荡30 min。(4)加入60 μL 20% SDS,65 ℃水浴2 h,水浴过程中每20 min轻轻上下颠倒几次。(5)6 000 r·min-1离心10 min,取上清液,加入等体积氯仿∶异戊醇(24∶1),上下颠倒几次,8 000 r·min-1离心10 min,取上清液。(6)加入10 g·L-1 RNA酶10 μL,37 ℃水浴30 min。(7)再次加入等体积氯仿∶异戊醇(24∶1)进行抽提,8 000 r·min-1离心10 min。(8)取上清液加入0.6倍体积异丙醇沉淀1 h,离心30 min。(9)弃上清液,加入预冷的70%乙醇,12 000 r·min-1离心10 min,该步骤重复1次。(10)室温下风干后,加入约20 μL ddH2O,保存备用。
1.2.4 16S rDNA扩增 以提取的DNA为模板,使用引物27F(5’-AGAGTTTGATCCTGGCTCAG-3’)和1 492R(5’-CGGTTACCTTGTTACGAC TT-3’)扩增16S rDNA[6],扩增片段长度大约1 500 bp。PCR扩增反应体系(50 μL):1×PCR缓冲液(Takara),0.2 mmol dNTP,2.5 mmol MgCl2,正反向引物各0.2 μmol,rTaq DNA聚合酶(Takara)1.25 U,DNA模板1 μL。反应参数:94 ℃预变性1 min,94 ℃变性1 min,56 ℃退火1 min,72 ℃延伸1.5 min,30个循环,72 ℃延伸10 min。PCR产物用1%琼脂糖凝胶电泳检测。
1.2.5 序列提交 将所有序列提交到GenBank数据库,注册号为KP114466-KP114524。
1.3 高通量测序分析细菌遗传多样性
1.3.1 样品总DNA的提取 样品用0.22 μm孔径滤膜过滤,过滤后把滤膜剪碎,用PBS缓冲液将菌体清洗下来,DNA提取方法同上述1.2.3。用1%琼脂糖凝胶电泳检测提取的基因组DNA。
1.3.2 PCR扩增及454焦磷酸测序 将提取的样品总DNA送至上海美吉生物医药科技有限公司进行高通量测序分析(采用Genome Sequencer FLX 测序系统)。提取的DNA有2个样品浓度太低,无法继续进行高通量测序分析。因此,4个样品只有消融区雪和冰川融水得到了高通量测序的结果。用标签测序法选择16S rRNA的V1-V3区构建群落文库。16S rRNA扩增使用的正向引物为![]() 其中,下划线碱基为454 Life Sciences®引物B,斜体碱基为细菌通用引物27F,黑体为一个含有2个碱基的连接序列,插入到454引物B和27F之间,用来减少合成引物对PCR反应效率的影响。反向引物为
其中,下划线碱基为454 Life Sciences®引物B,斜体碱基为细菌通用引物27F,黑体为一个含有2个碱基的连接序列,插入到454引物B和27F之间,用来减少合成引物对PCR反应效率的影响。反向引物为![]() 带下划线碱基为 454 Life Sciences®引物A,斜体碱基为细菌通用引物533R。NNNNNNNN为8个特定碱基标签来标记每个PCR产物,其碱基序列如表1所示。
带下划线碱基为 454 Life Sciences®引物A,斜体碱基为细菌通用引物533R。NNNNNNNN为8个特定碱基标签来标记每个PCR产物,其碱基序列如表1所示。
表1 5’ 端反向引物标签
Table 1 5’ tagged PCR reversed primers

样品Samples5’端反向引物序列(5’-3’)5’terminalreverseprimersequences消融区雪SnowsamplesfromablationzoneTATGCACG冰川融水GlacialmeltwaterTCGCGCTA
PCR反应体系为:1×FastPfu Buffer,0.25 mmol dNTP 2 μL,0.1 μmol引物,TransStart FastPfu高保真酶(TransGen AP221-02)2.5 U,DNA 模板10 ng,总体积20 μL。PCR反应程序:95 ℃预变性2 min,95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s,25 个循环,72 ℃ 延伸5 min。保存于10 ℃。PCR仪为ABI GeneAmp® 9700型。将同一样本的PCR产物混合后用2%琼脂糖凝胶电泳检测。将符合目的片段长度的平行扩增产物合并,使用Tiangen Gel Purification Kit试剂盒进行纯化回收,利用NanoDrop超微量分光光度计测定纯化后产物浓度,将所有位点样品等浓度混合后上机测序。
1.3.3 数据的分析方法
1.3.3.1 测序数据统计及数据优化 根据barcode序列区分各个样品的测序数据,按照barcode标签完全匹配方式提取有效序列。使用Qiime[7]找出barcode标签序列前引物序列并去掉。再去掉小于200 bp的碱基、模糊碱基及单碱基重复区,同时检测PCR扩增中产生的嵌合体序列并去除。
1.3.3.2 OTU聚类 使用Uparse[8]方法进行OTU聚类,OTU中序列相似性设为97%,得到OTU的代表序列,使用usearch_global方法将优化序列map比对回OTU代表序列,得到各样品OTU丰度统计表。
1.3.3.3 分类学分析 采用RDP classifier贝叶斯[9]算法对OTU代表序列进行分类学分析,并在各个水平统计每个样品的群落组成。使用Silva[10]数据库进行比对。
1.3.3.4 Alpha丰富程度分析方法 采用非参数法(Nonparametric estimators Good)估算样品代表群落的盖度指数(Community coverage),利用mothur软件计算比较每个样品的 Shannon、Simpson、Chao1和ACE多样性指数。
1.3.3.5 稀释性曲线 对序列采用随机抽样的方法,以抽到的序列数与它们所能代表OTU的数目构建稀释性曲线[11]。利用Mothur[12]做稀疏分析,利用R语言工具制作曲线图。
1.3.3.6 群落结构组分图 使用统计学方法,观测样品在不同分类水平上的群落结构。通常使用较直观的饼图或柱状图等形式呈现[13]。基于taxa_summary文件夹中的数据表,利用R语言工具作图或在Excel中编辑作图。
2 结果与分析
2.1 细菌纯培养结果
根据恢复出的菌株菌落特征,筛选出58个菌株,用于16S rDNA基因序列分析。
2.1.1 门水平的群落构成 此冰川细菌由Actinobacteria、Proteobacteria、Bacteroidetes、Deinococcus-Thermus4个门组成,各自在不同样品中的相对丰度如表2所示。4个门以Proteobacteria所占比例最高,其次为Actinobacteria,Bacteroidetes所占比例次之,Deinococcus-Thermus位居最后。
表2 不同样品门水平的细菌类群相对丰度
Table 2 Relative abundance of bacterial community at phylum level in different samples

门Phylum相对丰度/%Relativeabundance高位点雪Highaltitudesnow消融区雪Snowfromablationzone冰川融水Glacialmeltwater河水WaterfromglacialriverActinobacteria0.0516.2634.3167.11Proteobacteria86.8667.6962.7530.51Bacteroidetes11.286.092.792.38 Deinococcus-Thermus1.819.960.150.00
2.1.2 属水平的群落构成 在属水平上,此冰川细菌由Pseudomonas、Janthinobacterium、Flavobacterium、Devosia、Duganella、Deinococcus、Cryobacterium、Rugamonas、Arthrobacter、Mycetocola、Massilia、Salinibacterium组成(表3)。
表3 不同样品属水平上主要细菌类群的相对丰度
Table 3 Relative abundance of bacterial community at genus level in different snow and water samples
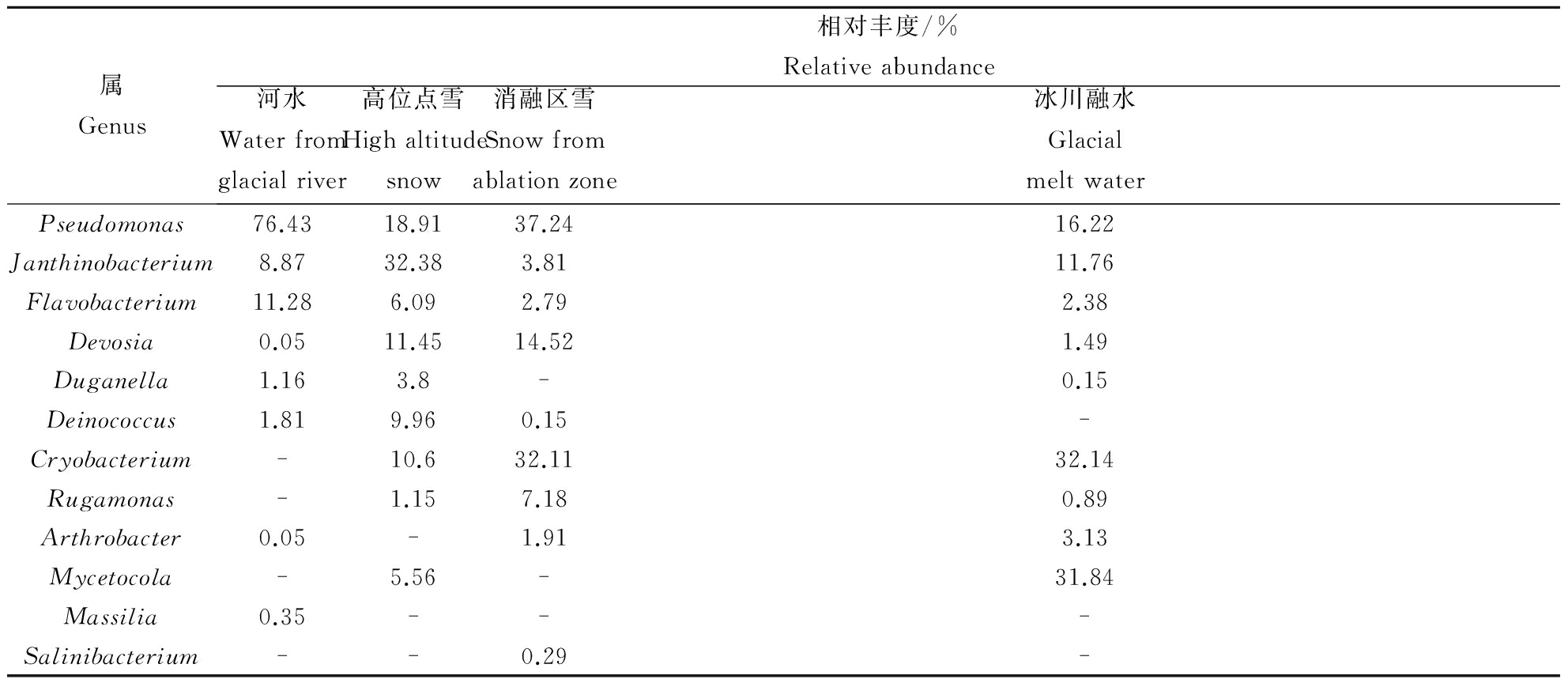
属Genus相对丰度/%Relativeabundance高位点雪Highaltitudesnow消融区雪Snowfromablationzone冰川融水Glacialmeltwater河水WaterfromglacialriverPseudomonas76.4318.9137.2416.22Janthinobacterium8.8732.383.8111.76Flavobacterium11.286.092.792.38Devosia0.0511.4514.521.49Duganella1.163.8-0.15Deinococcus1.819.960.15-Cryobacterium-10.632.1132.14Rugamonas-1.157.180.89Arthrobacter0.05-1.913.13Mycetocola-5.56-31.84Massilia0.35---Salinibacterium--0.29-
2.2 高通量测序结果
2.2.1 有效序列提取 各样品有效序列和优化序
列统计见表4。2个样品经454高通量测序共得到20 280条有效序列,序列平均长度为449.4 bp。对结果进行去杂,共得到11 444条优质序列,序列平均长度为402 bp。表4同时显示,平均优化效率为56.16%。优化序列用于样品间细菌丰富度和多样性的评估。
表4 各样品序列数据统计
Table 4 Statistics of sample sequence

样品Samples有效序列Validsequences优化序列Trimmedsequences优化:有效/%Valid/Trimmed消融区雪 Snowofablationzone10749652060.66冰川融水Glaciameltwater9531492451.66总计Totalsequences2028011444-
2.2.2 样品取样深度 本研究在0.97的相似度水平上绘制样品的稀释曲线,结果如图1。当序列较少时,随着样本中测序数量的增加,OTU数目剧增.随着测序量的不断增大,OTU数目增加幅度趋于平缓,说明本次试验的测序量基本能够反映样品中细菌群落的多样性组成。
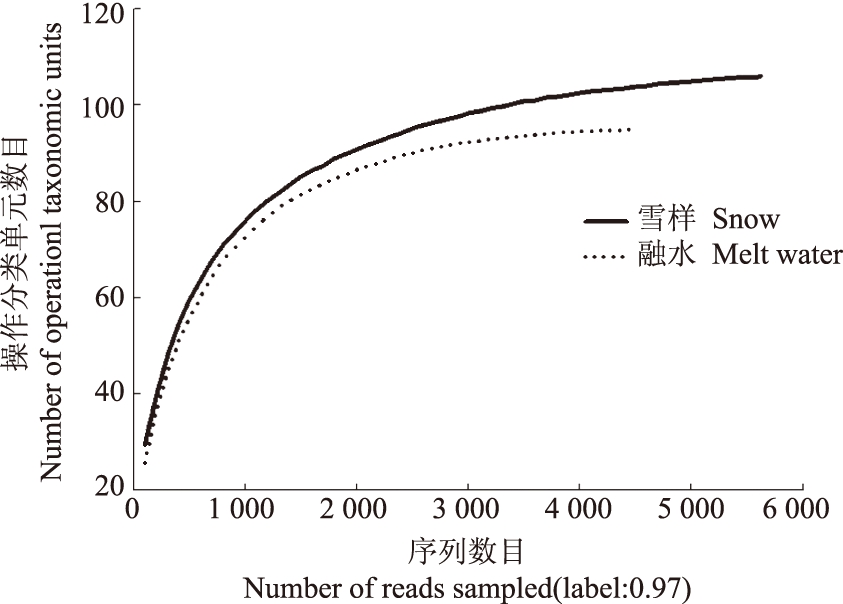
图1 样品稀释性曲线
Fig.1 Rarefaction cures of different samples
2.2.3 多样性指数分析 消融区雪和融水样得到的reads、OTU丰度、ACE、Chao指数、Shannon多样性指数、Simpson指数如表5所示。图1的稀释性曲线以及表5的多个指标的结果都说明消融区雪的细菌多样性大于冰川融水的多样性。
表5 不同样品物种多样性和丰富度估计
Table 5 Species diversity and abundance estimation
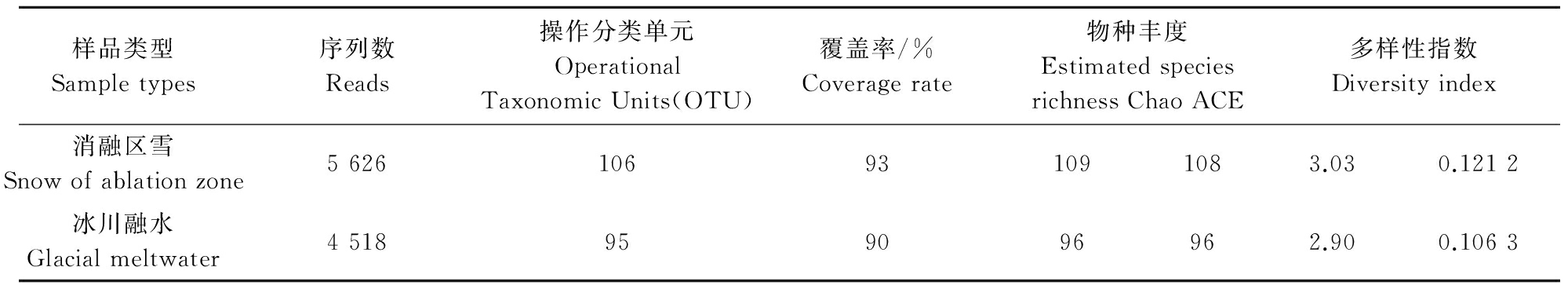
样品类型Sampletypes序列数Reads操作分类单元OperationalTaxonomicUnits(OTU)覆盖率/%Coveragerate物种丰度EstimatedspeciesrichnessChaoACE多样性指数Diversityindex消融区雪Snowofablationzone5626106931091083.030.1212冰川融水Glacialmeltwater4518959096962.900.1063
2.2.4 Venn图 如图2所示,消融区雪所特有的OTU为39,冰川融水所特有的OUT为28。二者共有的OTU为67,占消融区雪总OTU的62%,冰川融水总OTU的70.5%。共有OTU数在各自样品中所占比例较大,说明2种样品组成较相似。
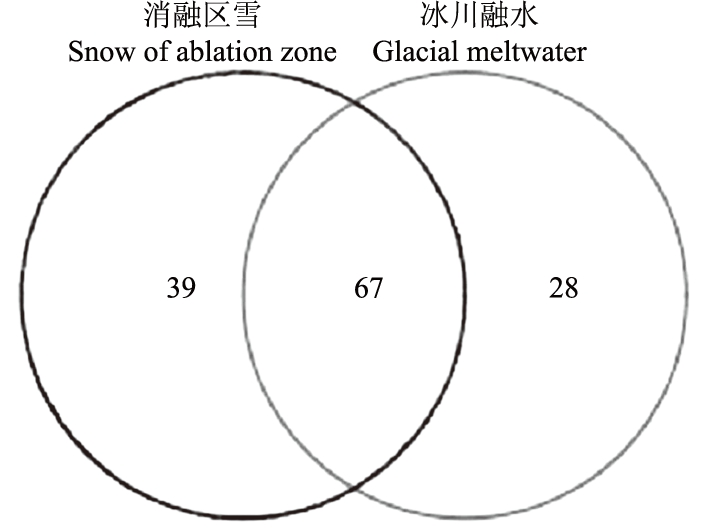
图2 消融区雪和冰川融水OTU分布Venn图
Fig.2 Number of unique and shared OTU between snow of ablation zone and glacial meltwater
2.2.5 门水平的群落构成 经454高通量测序分析,2个样品共检测到细菌的11个门:Acidobacteria、Actinobacteria、Bacteroidetes、Candidate division TM7、Chloroflexi、Cyanobacteria、Firmicutes、Gemmatimonadetes、Planctomycetes、Proteobacteria、Verrucomicrobia,主要由Proteobacteria、Bacteroidetes、Firmicutes这3种组成(表6)。其中Proteobacteria为2种样品的优势类群,在消融区雪所占比例为88.13%,而在冰川融水中为93.47%。Bacteroidetes在消融区雪和冰川融水所占比例分别为8.75%和4.87%,Firmicutetes在消融区雪和冰川融水所占比例分别为1.19%和1.31%,其他类群在2个样品中所占比例均小于1%。
表6 各样品在主要门分类水平上的菌群组成及其相对丰度
Table 6 Taxonomy information of dominant bacteria at phylum level and their relative abundance
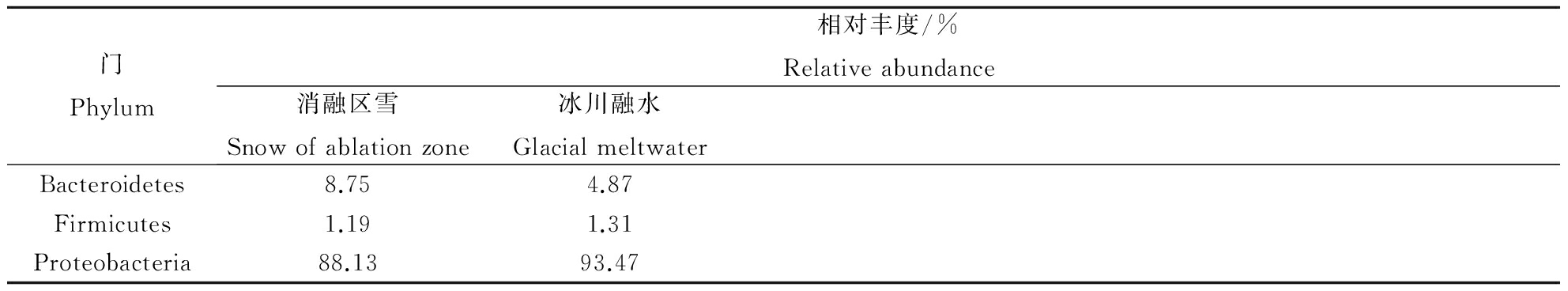
门Phylum相对丰度/%Relativeabundance消融区雪Snowofablationzone冰川融水GlacialmeltwaterBacteroidetes8.754.87Firmicutes1.191.31Proteobacteria88.1393.47
2.2.6 属水平的群落构成 在属的分布上(图3),2个样品中至少1个样品所占比例为1%以上的有Acidovorax、Bacillus、Bacteroides、Brevundimonas、Herbaspirillum、Massilia、Pedobacter、Phenylobacterium、Polaromonas、Pseudorhodobacter、Variovorax 11个属。
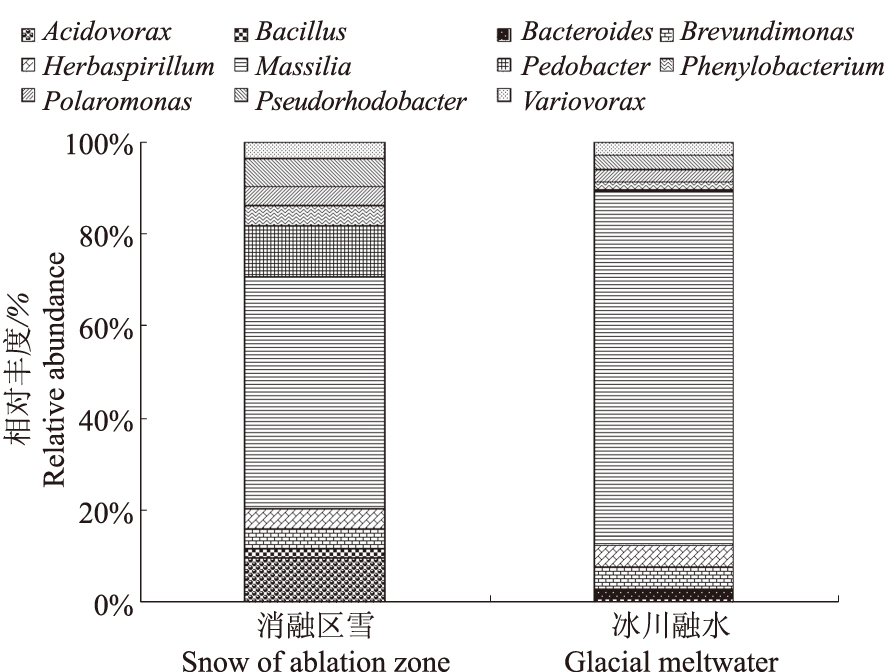
图3 基于高通量测序的细菌属水平的群落结构
Fig.3 Bacterial community distribution at genus levels based on high-throughput sequencing
3 讨论
在本研究中,高通量测序的结果是Massilia在雪样中的比例比在冰川融水中的低,而在老虎沟12号冰川,通过培养方法得到的结果是Massilia在雪样中的比例比在土样中的高[2]。存在这种差异的原因,一方面可能是由于研究方法的差异,另一方面也可能是研究位点的差异。如在天山1号冰川,也用了高通量测序,但未检测到该属[14]。而在玉珠峰,Shen等不仅检测到了该属,而且还对该属进行了新种的鉴定[15]。
在门水平上,纯培养和高通量测序同时检测到了大量的Proteobacteria和Bacteroidetes。但每种方法都检测到了另一种方法所未检测的细菌群落。如纯培养检测到了大量的Deinococcus-Thermus,而高通量测序则检测到了大量的Firmicutes。在属水平上,除了Massilia、Cryobacterium、Devosia、Flavobacterium、Pseudomonas 5种属被2种方法同时检测到以外,其它的属仅被1种方法所分析到。HUANG等[16]使用标记编码FLX扩增子焦磷酸测序(Bacterial Tag Encoded FLX Amplicon Pyrosequencing,bTEFAP)、克隆文库构建、纯培养3种方法对南极Tawani(P)湖的细菌多样性进行研究,结果表明使用多种方法研究比任何一种单一的方法能获得更全面的细菌群落信息。这一结果与本研究的结果相一致。因此本研究将2种方法所得到的结果综合起来,用于分析熊彩岗日冰川的细菌群落多样性。
2种方法获得的所有属水平的细菌与其他多个位点的冰川进行比较,如通过高通量测序分析的天山1号冰川[14]、High Arctic[17]、南极的Tawani湖[16]、老虎沟12号冰川[3],通过纯培养以及克隆文库构建等方法研究的东绒布冰川[18-19]、马兰冰川[20-21]、Palong[18]、 Kafni冰川土样[22]、老虎沟雪坑样品[23]、GISP 2[24]、南极Terra Nova海湾的水柱[25]、敦德冰芯及慕士塔格冰芯[1]。仍有上述这些研究中尚未见到的细菌资源,如:高通量测序检测到的Arcobacter、Arcticibacter、Blastocatella、Blautia、Paraprevotella、Phascolarctobacterium、Pseudofulvimonas、Subdoligranulum,培养方法获得的Mycetocola、Rugamonas。因此,增加研究位点,对于更好的了解地球上冰川细菌的多样性很有必要[17]。
通过纯培养与高通量测序相结合的方法,对熊彩岗日地区冰川不同海拔形成的雪样和水样中的细菌多样性进行了分析。纯培养结果表明,Massilia只在雪样中存在,Salinibacterium只在水样中存在,其他属在雪样和水样中都存在,说明雪样和水样组成是比较相似的。图2中,消融区雪和冰川融水共有OTU数占各自样品的比例较大,Shannon指数也相近,也说明雪样和水样的组成是相似的。LAROSE等[26]对极地雪和融水的细菌的研究表明,雪和融水中群落组成具有很大差异。刘勇勤等[27]对Yala冰川雪、冰碛湖、冰川溪流3种生境细菌多样性的研究表明,冰川雪和水是高度多样化的系统。本研究结果与以上结果并不一致。上述2种研究使用克隆文库的方法,与本研究使用方法不同,同时研究位点也有差异,这些可能都是与本研究结果不一致的原因。
4 结论
本研究利用培养及高通量测序的方法对熊彩岗日地区冰川细菌多样性及分布特征进行了研究,培养结果表明,冰川细菌由Bacteroidetes、Actinobacteria、Deinococcus-Thermus、Proteobacteria 4个门组成,属水平由Pseudomonas、Janthinobacterium、Flavobacterium、Devosia、Duganella、Deinococcus、Cryobacterium、Rugamonas、Arthrobacter、Mycetocola、Massilia、Salinibacterium组成。高通量测序结果表明冰川细菌主要由Proteobacteria、Bacteroidetes、Firmicutes 3个门组成,由Acidovorax、Bacillus、Bacteroides、Brevundimonas、Herbaspirillum、Massilia、Pedobacter、Phenylobacterium、Polaromonas、Pseudorhodobacter、Variovorax等属组成。雪样和水样组成较相似。2种方法均检测到了熊彩岗日地区冰川所特有的细菌类群。
参考文献:
[1] 陈勇. 青藏高原冰雪微生物多样性及其与气候环境关系的研究[D]. 兰州: 兰州大学, 2009.
[2] 张淑红, 侯书贵, 秦翔, 等. 祁连山老虎沟12号冰川退缩对细菌优势种群影响的初步研究[J]. 冰川冻土, 2013, 35(3): 751-760.
[3] ZHANG S, HOU S, QIN X, et al. Preliminary study on effects of glacial retreat on the dominant glacial snow bacteria in Laohugou Glacier No. 12[J]. Geomicrobiology Journal, 2015, 32(2): 113-118.
[4] LI Z, FANG H, TIAN L, et al. Changes in the glacier extent and surface elevation in Xiongcaigangri region, Southern Karakoram Mountains, China[J]. Quaternary International, 2015, 371(12): 67-75.
[5] ZHOU J, BRUNS M A, TIEDJE J M. DNA recovery from soils of diverse composition [J]. Applied and Environmental Microbiology, 1996, 62(2): 461-468.
[6] CHAKRAVORTY S, HELB D, BURDAY M, et al. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria[J]. Journal of Microbiological Methods, 2007, 69(2): 330-339.
[7] CAPORASO J G, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods, 2010, 7(5): 335-336.
[8] EDGAR R C, HASS B J, CLEMENTE J C, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics, 2011, 27(16): 2194-2200.
[9] WANG Q, GARRITY G M, TIEDJEi J M, et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology, 2007, 73(16): 5261-5267.
[10] QUAST C, PRUESSE E, YILMAZ P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools[J]. Nucleic Acids Research, 2013, 41 (D1): D590-D596.
[11] AMATO K R, YEOMAN C J, KENT A, et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes[J]. The ISME Journal, 2013, 7(7): 1344-1353.
[12] SCHLOSS P D, GEVERS D, WEATCOTT S L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies[J]. PLoS One, 2011, 6(12): e27310.
[13] OBERAUNER L, ZACHOW C, LACKNER S, et al. The ignored diversity: complex bacterial communities in intensive care units revealed by 16S pyrosequencing[J]. Scientific Reports, 2013, 3(5-6): 413-1424.
[14] WU X, ZHANG W, LIU G, et al. Bacterial diversity in the foreland of the Tianshan Glacier No. 1, China[J]. Environmental Research Letters, 2012, 7(1): 1-9.
[15] SHEN L, LIU Y, WANG N, et al. Massilia yuzhufengensis, isolated from an ice core[J]. International Journal of Systematic and Evolutionary Microbiology, 2012, 64(4): 1285-1290.
[16] HUANG J P, SWAIN A K, THACKER R W, et al. Bacterial diversity of the rock-water interface in an East Antarctic freshwater ecosystem, Lake Tawani(P)+[J]. Aquatic Biosystems, 2013, 9: 4. doi:10.1186/2046-9063-9-4.
[17] SCHÜTTE U M, ABDO Z, FOSTER J, et al. Bacterial diversity in a glacier foreland of the high Arctic[J]. Molecular Ecology, 2010, 19 (Suppl. 1): 54-66.
[18] LIU Y, YAO T, JIAO N, et al. Bacterial diversity in the snow over Tibetan Plateau Glaciers[J]. Extremophiles, 2009, 13(3): 411-423.
[19] ZHANG S, HOU S, MA X, et al. Culturable bacteria in Himalayan glacial ice in response to atmospheric circulation[J]. Biogeosciences, 2007, 4: 1-9.
[20] XIANG S, YAO T, AN L, et al. Change of bacterial community in the Malan ice core and its relation to climate and environment[J]. Chinese Science Bulletin, 2004, 49(17): 1869-1875.
[21] XIANG S, YAO T, AN L, et al. Bacterial diversity in Malan ice core from the Tibetan Plateau[J]. Folia Microbiologica, 2004, 49(3): 269-275.
[22] SRINIVAS T N, SINGH S M, PRADHAN S, et al. Comparison of bacterial diversity in proglacial soil from Kafni Glacier, Himalayan Mountain ranges, India, with the bacterial diversity of other glaciers in the world[J]. Extremophiles, 2011, 15(6): 673-690.
[23] ZHANG S, YANG G, WANG Y, et al. Abundance and community of snow bacteria from three glaciers in the Tibetan Plateau[J]. Journal of Environmental Sciences, 2010, 22(9): 1418-1424.
[24] MITEVA V I, SHERIDAN P P, BRENCHLEY J E. Phylogenetic and physiological diversity of microorganisms isolated from a deep Greenland glacier ice core[J]. Applied and Environmental Microbiology, 2004, 70(1): 202-213.
[25] GIUDICE A L, CARUSO C, MANGANO S, et al. Marine bacterioplankton diversity and community composition in an Antarctic coastal environment[J]. Microbial Ecology, 2012, 63(1): 210-223.
[26] LAROSE C, BERGER S, FERRARI C, et al. Microbial sequences retrieved from environmental samples from seasonal Arctic snow and meltwater from Svalbard, Norway[J]. Extremophiles, 2010, 14(2): 205-212.
[27] LIU Y, YAO T, JIAO N, et al. Microbial diversity in the snow, a moraine lake and a stream in Himalayan glacier[J]. Extremophiles, 2011, 15(3): 411-421.
(责任编辑:朱秀英)
Study of bacterial diversity in glaciers of Xiongcaigangri region of south Karakoram
Abstract: Using the methods of pure culture and high-throughput sequencing, bacteria diversity was investigated in glacial snow and glacial water from Xiongcaigangri region of south Karakoram. Pure culture results showed that glacial bacteria were composed of Proteobacteria, Bacteroidetes, Actinobacteria, Deinococcus-Thermus at phylum level. They were composed of Pseudomonas, Janthinobacterium, Flavobacterium, Devosia, Duganella, Deinococcus, Cryobacterium, Rugamonas, Arthrobacter, Mycetocola, Massilia, Salinibacterium at genus level. High-throughput sequencing results showed that glacial bacteria were mainly composed of Proteobacteria, Bacteroidetes, Firmicutes at phylum level. They were mainly composed of Acidovorax, Bacillus, Bacteroides, Brevundimonas, Herbaspirillum, Massilia, Pedobacter, Phenylobacterium, Polaromonas, Pseudorhodobacter, Variovorax at genus level. The results showed that the combination of culture-dependent method and high-throughput sequencing could obtain more comprehensive investigation of bacterial community. From the two methods, bacterial diversity was similar between glacial snow and glacial water, while with some unique community. In the meantime, some unique bacteria were detected in glacial environments of Xiongcaigangri region.
Key words: bacteria; diversity; glacier; Xiongcaigangri region
中图分类号:Q 938
文献标志码:A
收稿日期:2015-04-06
基金项目:国家自然科学基金项目(31100369,41101072,41330526)
文章编号:1000-2340(2016)02-0275-07